Deploy and build with foundation models for biology — across histology, transcriptomics, and genomics.
Bioptimus builds foundation models that learn the dynamics of human biology across scales — from cell to tissue to organ. This documentation covers how to access, deploy, and build with our models on AWS and on-premise.
Bioptimus models are pretrained on large, diverse histology data, so their representations transfer to many tasks without training from scratch. Point the SDK at a slide and a model, and it tiles the slide, masks out background, runs the model, and writes results to disk. For precise definitions, see the Glossary.
1. Whole slide image
2. Tissue segmentation
3. Tile embeddings
4. Spatial gene expression
A scanned H&E slide can be billions of pixels — too large to process at once — so the pipeline starts from the slide and splits it into small tiles, each processed independently.
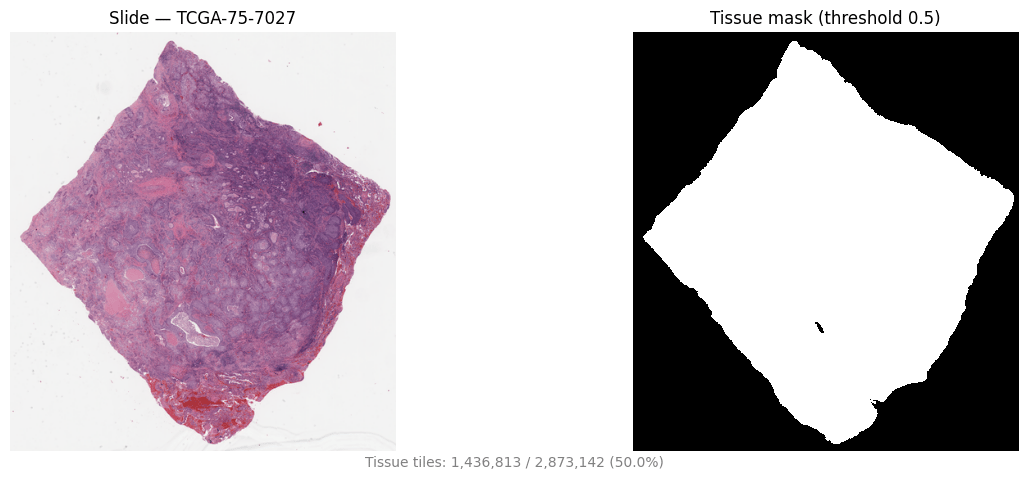
Slides are mostly background. The pipeline tiles coarsely (512×512 at 8 µm/px) and runs tissue segmentation to produce a tissue map, keeping only tissue-bearing tiles — cutting cost before the expensive feature step.
Tissue mask (right) beside the slide thumbnail (left), TCGA-LUAD TCGA-75-7027. Background is discarded before feature extraction. Representative example.
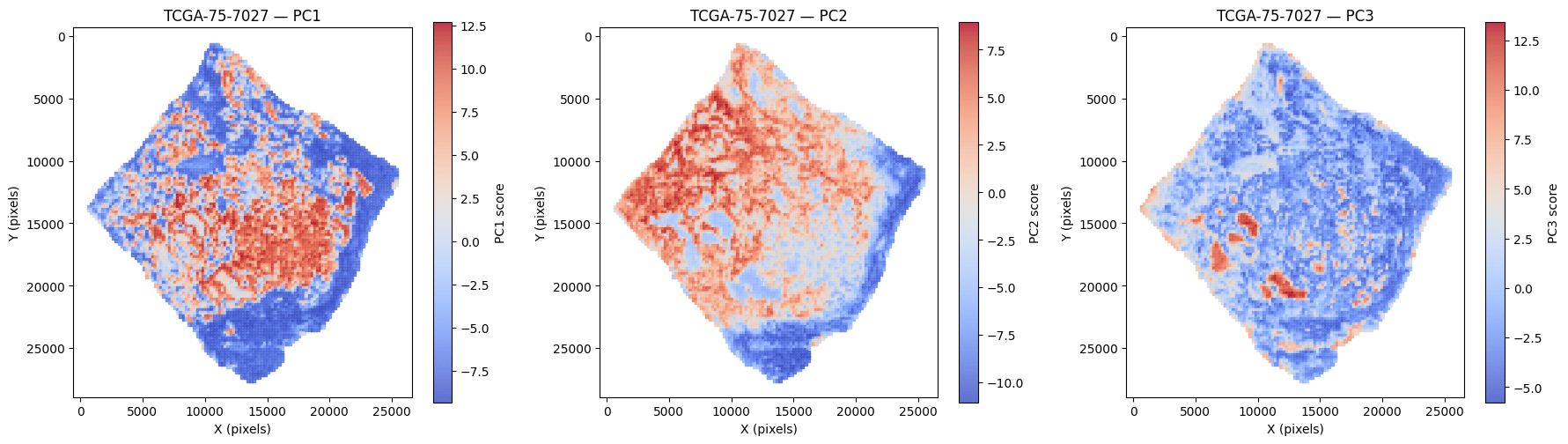
Tissue tiles (224×224 at 0.5 µm/px) are embedded by H-Optimus into a 1536-d feature vector each. Principal components of these embeddings reveal the dominant axes of morphology — tumor, stroma, and immune compartments.
Principal components of tile embeddings mapped back onto the slide (TCGA-LUAD TCGA-75-7027) — PC1–PC3 each isolate a distinct tissue region. Representative example.
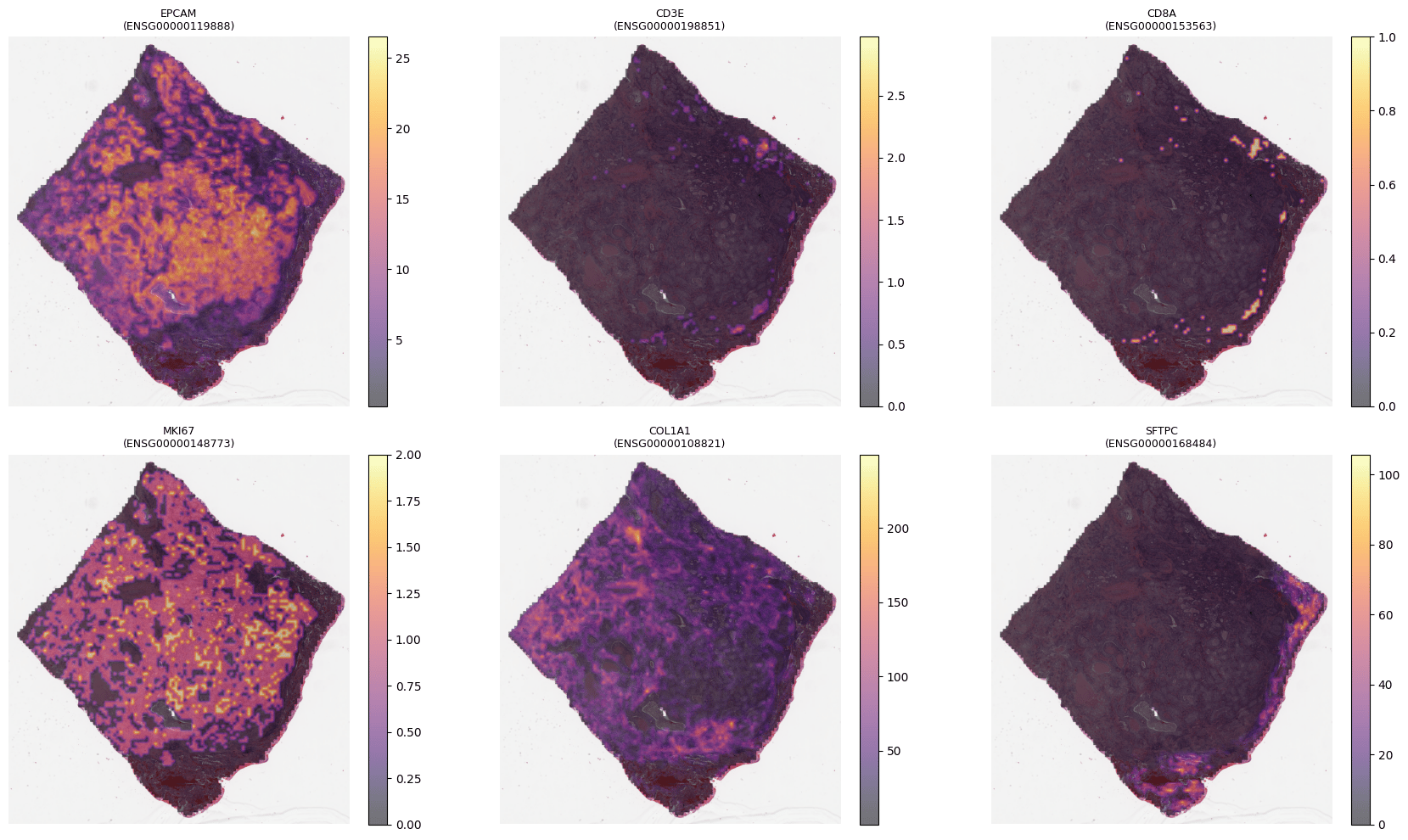
M-Optimus produces the same 1536-d embeddings and predicts spatial gene expression directly from each tile (optionally informed by bulk RNA) — a molecular readout without a spatial assay.
Spatial expression for a six-gene panel predicted from H&E (M-Optimus, TCGA-LUAD TCGA-75-7027). Representative example.